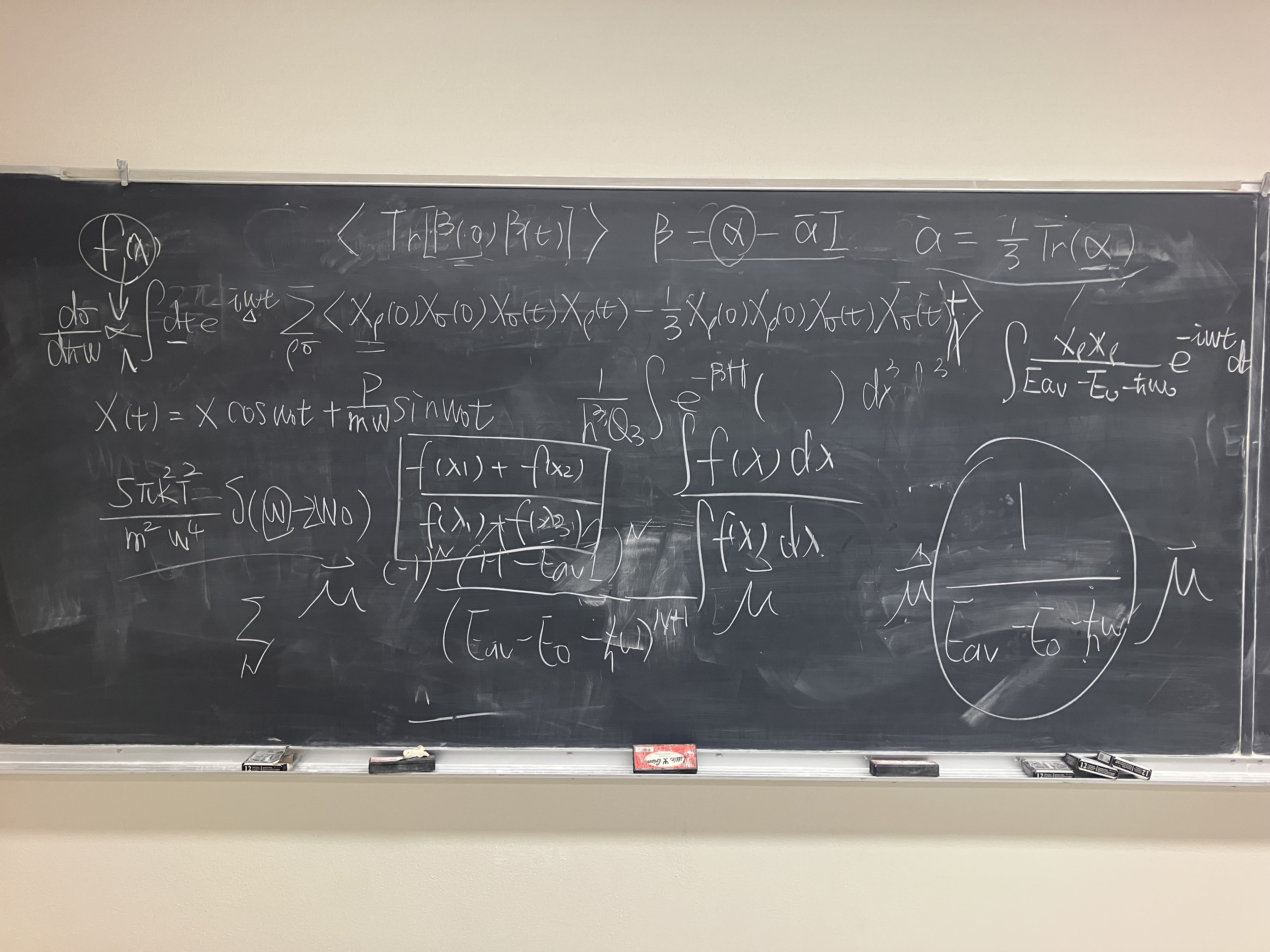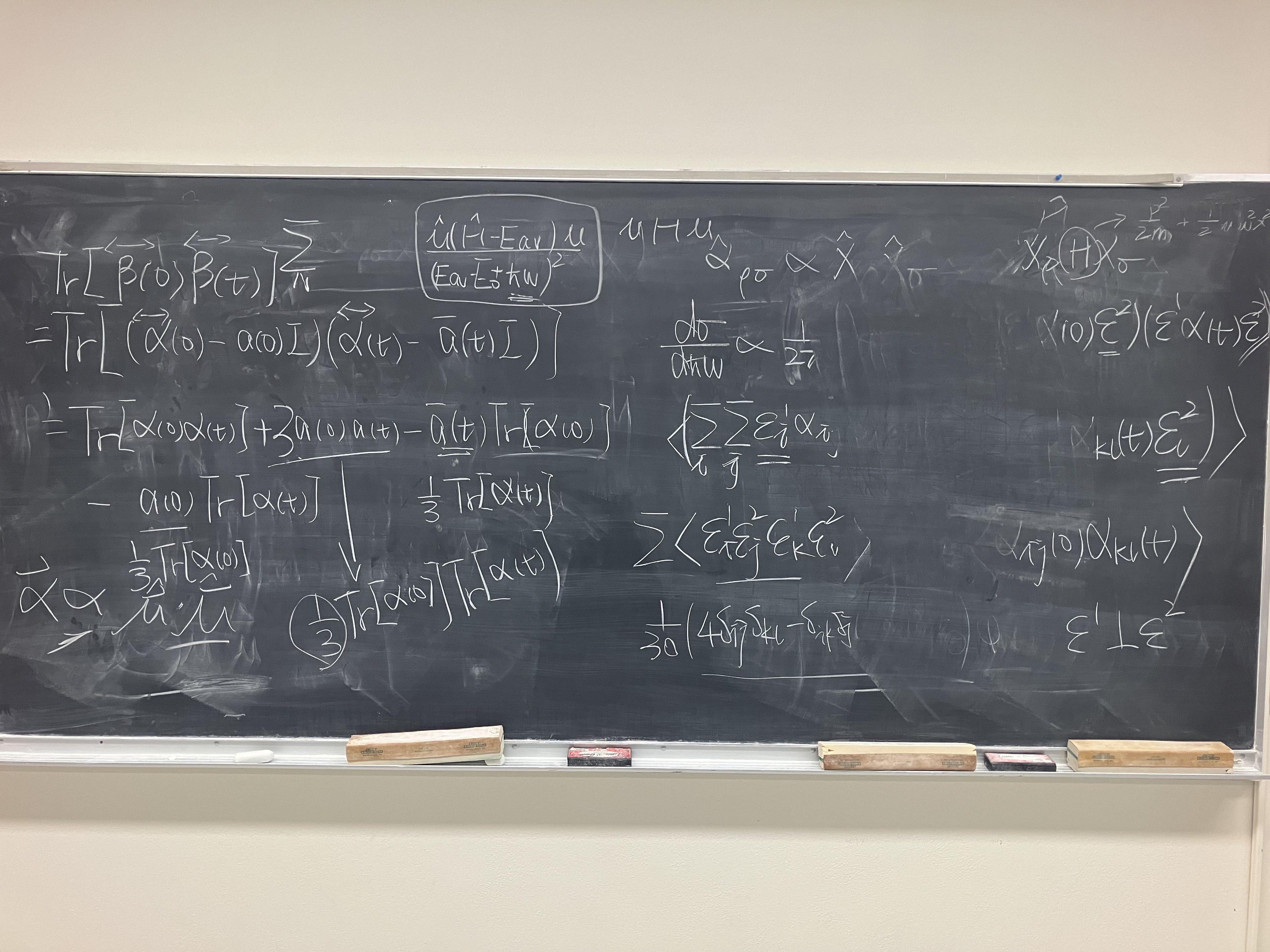When a system of molecules is placed in an electric field, the electrons and nuclei will be displaced in such a manner as to induce dipole moments. If we make the valid assumption that the electric field is weak, the induced dipole moment will be proportional to the field strength. The proportionality coefficient is called the polarizability, α. Then in 3D space, the induced dipole moment is given by
μ=α⋅E
which is the dot product of a 3×3 tensor and a 3 dimentional column vector. If E=E0cosωt, then μ will vary with time according to
μ=α⋅E0cosωt
However, in real situations the vibration and the rotation of the molecule should be taken into account, which means that the polarizability is a function of time. For simplicity, let these be diatomic molecules and take small oscillation approximation:
α=α0+α1cosω0t
in which ω0 denotes for the frequency of the normal mode and α1 is a measure of how the polarizability varies with vibration.
This equation shows us that the induced dipole oscillates not only with the incedent frequency ω, but also with the frequency ω±ω0. The first term accounts the Rayleigh scattering, and the second and third terms are called the Stokes and anti-Stokes scattering, respectively. We see that the frequencies observed in Raman scattering are beat frequencies between the radiation frequency ω and the molecular frequency ω0, and the frequency difference between the incident light and the scattered light is called the Raman shift.
Classical Electrodynamics tells us that the mean rate of radiation emitted over all directions by a dipole oscillating according to μ=μ0cosωt is given by
I=3c3ω4∣μ0∣2
where c is the speed of light. The intensity of the scattered light is proportional to the square of the amplitude of the induced dipole moment, so there exist the proportionality where
IRaman∝∣α1⋅E0∣2=E0T⋅α1T⋅α1⋅E0
and if some proper approximation is made (as we shall do later in quantum description of Raman scattering), the polarizability tensor could be symetric, thus we have
IRaman∝E0T⋅α12⋅E0
Notes that α1 stands for the small change of polarizability due to the vibration, so one can easily understand that when doing static calculation people always use the value ∣∂α/∂Q∣2 at the equilibrium geometry, where Q is the normal mode coordinate, to obtain the full spectroscopy. This was fully discussed in the 5th chapter of THE RAMAN EFFECT, 2002 by Derek A. Long.
Classical theory is not enough for us to understand the process of photon scattering by non relativistic atomic electrons. Concepts discussed above is just a rough physical picture of the Raman scattering process.
Quantum Theory of Raman Scattering
Kramers-Heisenberg Formula
With the help of Quantum Field Theory, one can treat the scattering process as creation and annihilation of photons and consider photons as quantum-mechanical excitations of the radiation field. The interaction Hamiltonian between the atomic electrons and the radation field is given by
where the summation is over the various atomic electrons that participate in the interaction, and the expression A(xi,t) originates from the vector potential and is now a field operator assumed to act on a photon state or a many-body state at xi, where xi is the position operator of the ith electron.
Recalling the Time-Dependent Second-Order Perturbation Theory, the transition probability from an initial state ∣i⟩ to a final state ∣f⟩ is given by
If we describe the incedent light as with (ℏω,ϵ) where ϵ is the unit vector of the incoming direction and also the scattered light (ℏω′,ϵ′) where ϵ′ is the unit vector of outcoming direction, after some fancy derivation in J. J. Sakurai’s book ADVANCED QUANTUM MECHANICS, 1967, the transition probability in some solid angle can be written as
This is called the Kramers-Heisenberg formula. It was derived before the advent of quantum mechanics by Hendrik Kramers and Werner Heisenberg in 1925, based on the correspondence principle applied to the classical dispersion formula for light. The quantum mechanical derivation was given by Paul Dirac in 1927. When the final state and the initial state do not coincide (as always the case in Raman scattering), the first term in the square bracket vanishes, and because the scattered frequency is usually close to the incident frequency, ω≃ω′, we may write the differential corss section of Raman scattreing as
This could be the starting point of introducing the correlation function formalism for AIMD simulation, but there exists one main obstacle: the dependence on n in the denominator of α^. So we have to do some simplification to make the formula more tractable.
Simplification for the Polarizability
Learned from the book RAMAN SPECTROSCOPY: THEORY AND PRACTICE by Herman A. Szymanski, this siplification is straight forward, involving a series expansion and a truncation. Let’s take out the first term
α^1=n∑En−Ei−ℏωμ^∣n⟩⟨n∣μ^
as an example and later we will find that this mathematical trick suits the second term α^2 as well.
Set the initial state always be the ground state, so we substitude E0 for Ei. I claim that there EXIST some constant "average energy" Eav such that the following equation holds:
Note that this series is not only a geometric series but also alternating series, so it converges pretty fast. If we truncate the series at the first term when N=0 and assume the polarizability does not change too much when light is shed on the molecule (which means this is the zero-order approximation), we have the following proportionality:
α^1∝μ^μ^orα^1,ρσ∝μ^ρμ^σ
where ρ,σ run from 1 to 3 denoting for x,y,z in 3D space.
Obviously, this mathematical trick is not only applicable to the first term α^1, but also to the second term α^2. Put them together, what really matters is the following proportionality:
α^∝μ^μ^∝x^x^orα^ρσ∝μ^ρμ^σ∝x^ρx^σ
By the elegance of proportionality, I have eliminated the somehow weird Eav. The EXISTENCE of Eav is all I need! Go back, we may now introduce our correlation function formalism.
Correlation Function formalism
This part follows Chapter 21: The Time-Correlation Function Formalism I, STATISTICAL MECHANICS, 1976 by Donald A. McQuarrie. Do ensemble average to the differential cross section and switch the delta function to the time domain, we have
where ρi is the density distribution of the initial state. It is pretty easy to see that the summation over i,f gives ensemble average, and handing out the eiωft,e−iωit paves the way to Dirac interaction picture, which gives
dℏωddΩdσ∝∫−∞∞dte−iωt⟨(ϵ⋅α^(0)⋅ϵ′)(ϵ⋅α^(t)⋅ϵ′)⟩
where (ϵ⋅α^(0)⋅ϵ′)(ϵ⋅α^(t)⋅ϵ′) can be written in component form as
where ⟨ϵiϵj′ϵkϵl′⟩sphere is the average over the solid angle. In experiments, people always put the observer perpendicular to the incident light beam, so we set ϵ⊥ϵ′, thus we have
In practice, because quantum correlation functions are usually difficult to calculate, classical autocorrelation functions obtained from classical simulations are more often used for spectra computation. Within the harmonic oscillator model, the Fourier transform of quantum and classical autocorrelation function only differ by a frequency-dependent prefactor, as we have already seen in infrared spectroscopy.
Although the idea for calculating both classical and quantum average is quite simple, as is mentioned in Yiwen’s note for IR spectrum, summation over ρ,σ in Raman spectrum brings more difficulties above. Introducing 3D isotropic harmonic approximation:
H=2mp12+p22+p32+21mω02(x12+x22+x32)
thanks to Wolfram Mathematica 13.0.0, Sthdent Edition, the classical average takes
The Mathematica .nb file is attached as appendix. Please contact me if
you find any mistakes.
Conclusion
In this note, I start with the classical theory for light scattering, giving a physical picture of this process and then terned to quantum description. The Kramers-Heisenberg Formula comes from the Time-Dependent Second-Order Perturbation Theory which focuses on the transition probability of a system under harmonic perturbation. As the transition probability is proportional to the intensity of scattered light, we then jump to the expression of scattering differential cross section, where we begin the rest of our journey.
The correlation function formalism cannot be introduced directly because the summation over states in the oringial formula. Under zero-th-order approximation, we let some “average energy” of the states to replace the summation over states, and then siplification of the polarizability leads to the proportionality between the polarizability tensor and the summation and multiplication of position operators, which is further calculated by mathematical software.
One thing I have to emphasize is that the truncation of our polarizability and later the introduction of proportionality together get rid of the frequency dependence (ω in the denominator) of the original polarizability. This only holds when the polarizability does not change too much during the measurement. \textcolor{RoyalBlue}{\textbf{In some situations, this could have some influence on the final prefactor}}. Let fq(λ) and fc(λ) and stands for the frequency dependence of the polarizability, then the prefactor could look like